
After the host is infected by the virus, the immune system can generate a corresponding immune response. Antibodies specific to viral surface proteins produced at this time can often prevent the virus from adhering to the surface of host cells, making it incapable of infecting cells. However, in some cases, antibodies play the opposite role during viral infection, they assist the virus to enter the target cells and increase the infection rate, this phenomenon is antibody-dependent enhancement (ADE).
The role of ADE was first reported by Hawkes in the 1960s. Since then, the presence of ADE has been found in the infection of more than 40 kinds of viruses in multiple families and genera. It is often difficult to prevent and control such viral diseases with ADE effects.
Mechanism of ADE Action in Viral Infection
This intricate mechanism has been the focus of intense scrutiny, with scientists examining the involvement of various immune cells such as macrophages, monocytes, B cells, neutrophils, and granulocytes. Further studies have confirmed the involvement of FcRII-specific monoclonal antibodies in blocking ADE when West Nile virus infected macrophage-like cell lines.
Another intricate mechanism of ADE action in flavivirus infection is ADE action mediated by complement receptor (CR). Researchers discovered that the infectivity of West Nile virus to P388D1 cells with FcR was enhanced in the presence of antiviral IgM, and that the specific monoclonal antibody of complement type 3 receptor (CR3) could block this enhancement. Interestingly, FcR antibodies could not block this effect, adding another layer of complexity to the ADE action mechanism.
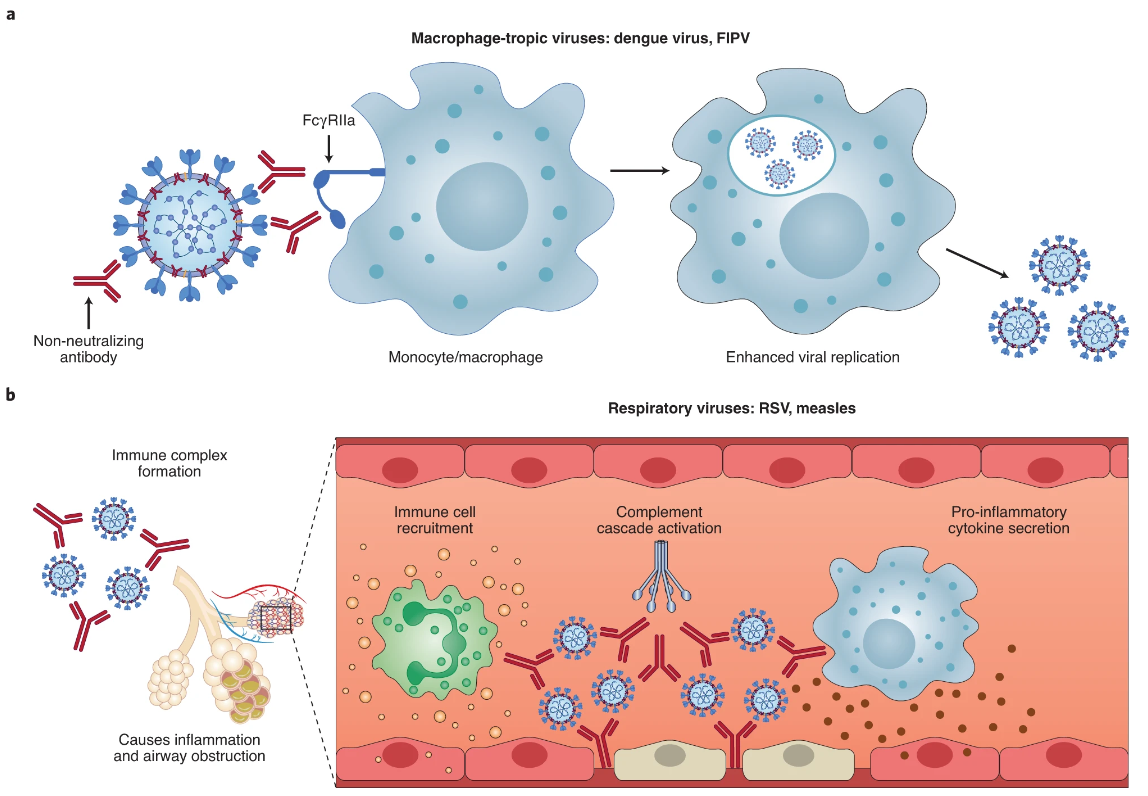
Figure 1. Two main ADE mechanisms in viral disease.
In vitro studies of human immunodeficiency virus infection have revealed at least five perplexing mechanisms of the ADE effect. These include: the interaction between the virus-antibody complex and FcR, which enhances adhesion to cells; the virus-antibody-complement complex interacts with the CR2 on the target cell, enhancing adhesion; the deposition of complement components on virions assists in the fusion of the viral envelope and the cell membrane; through FcR or CR, complement activation products and intracellular signal transduction have a stimulating effect on target cells, such as enhancing cellular endocytosis; and when the antibody or soluble CD4 is below the neutralizing concentration, it can bind to one subunit of the gp120 oligomer, causing changes in the configuration of other subunits, activating gp120, and promoting the fusion of the viral envelope and the cell membrane.
Despite the perplexing nature of the ADE action mechanism, it is crucial to continue studying it to develop effective treatments for viral infections. With its intricate and mind-bending complexities, ADE action mechanism remains a captivating topic for researchers to explore further.
Significance of Research on the Mechanism of Action of ADE
The salience of scrutinizing the intricacies of antibody-dependent enhancement (ADE) is rooted in its ability to exacerbate diseases and catalyze outbreaks. A prime example of this was Thailand’s horrific dengue virus infection epidemic in the 1980s, whereby young children who had previously contracted the virus often exhibited dengue hemorrhagic fever and dengue shock syndrome. Moreover, these children frequently underwent secondary infections, predominantly involving a blend of dengue virus types 1 and 2. Subsequently, researchers discovered that pre-infection with dengue virus type 4 in monkeys could intensify subsequent infections with dengue virus type 2, and inoculating immune serum into these primates could instigate high levels of viremia. Collectively, these findings revealed that while the emergence of dengue hemorrhagic fever and shock syndrome is determined by numerous factors, ADE can indeed augment the pathogenicity of dengue virus infection.
Ever since the advent of the rabies vaccine by Pasteur, it has been discovered that pre-existing antibodies can expedite disease progression and aggravate disease severity in various viruses. Although this phenomenon is not necessarily entirely attributable to ADE, it is still a formidable concern for researchers studying antiviral vaccines. As evidenced by numerous studies, the inactivated vaccine for goat arthritis not only fails to deter infection, but also fosters the occurrence of inflammatory diseases. At times, vaccines for monkey AIDS and equine infectious anemia can also incite infection. In clinical practice, vaccination with the porcine reproductive and respiratory syndrome virus (PRRSV) vaccine can occasionally exacerbate symptoms. Antibodies elicited by the PRRSV vaccine virus can heighten the replication of wild strains of the virus in pigs, and the propagation of the vaccine virus can also be augmented by maternal antibodies engendered by wild strains of the virus. Following a pig population’s infection with wild strains of PRRSV, the levels of immunity within the population fluctuate dramatically, and there are marked disparities in ADE between different individuals. Additionally, piglets that are passively immunized against PRRSV by maternal antibodies exhibit ADE activity when the antibody levels drop below protective levels (sub-neutralizing levels), which escalates the risk of animal infection and disease.
Significance of Research on the Mechanism of Action of ADE
The genetic diversity of viruses is prodigious, and this diversity is correlated with the development of vaccines, as immunity provoked by one strain may only partially resist limited infections by different strains. The virus strain displays a vast array of antigenic diversity, and there are significant disparities in ADE. Therefore, it is imperative to employ pathological models to scrutinize the position and role of ADE in the pathogenic mechanism of viruses with ADE effects, identify the antigenic determinants linked to ADE in these viruses, and modify or eliminate them from candidate vaccine viruses in order to engender safe and effective vaccines.