
Reprogramming of lipid metabolism has become an important feature of cancer. In order to adapt to the hypoxic and nutrient-deficient microenvironment, tumor cells, in addition to increasing glucose uptake and aerobic glycolysis, also need to undergo lipid metabolism reprogramming to enhance their biological behaviors. It is characterized by increased lipid uptake, lipid synthesis, fatty acid oxidation (FAO), and lipid storage. Increasing evidence shows that lipids serve as energy sources, membrane structures, signaling molecules (including biologically active lipids such as S1P, PGE2, and LPA), and even cause epigenetic modifications through fatty acylation of key molecules. Play a key role in cancer progression. Mechanistically, alterations in the lipid metabolism phenotype of tumor cells are directly driven by ongoing oncogenic events and extracellular tumor microenvironment (TME) factors such as hypoxia, acidosis, and nutritional alterations.
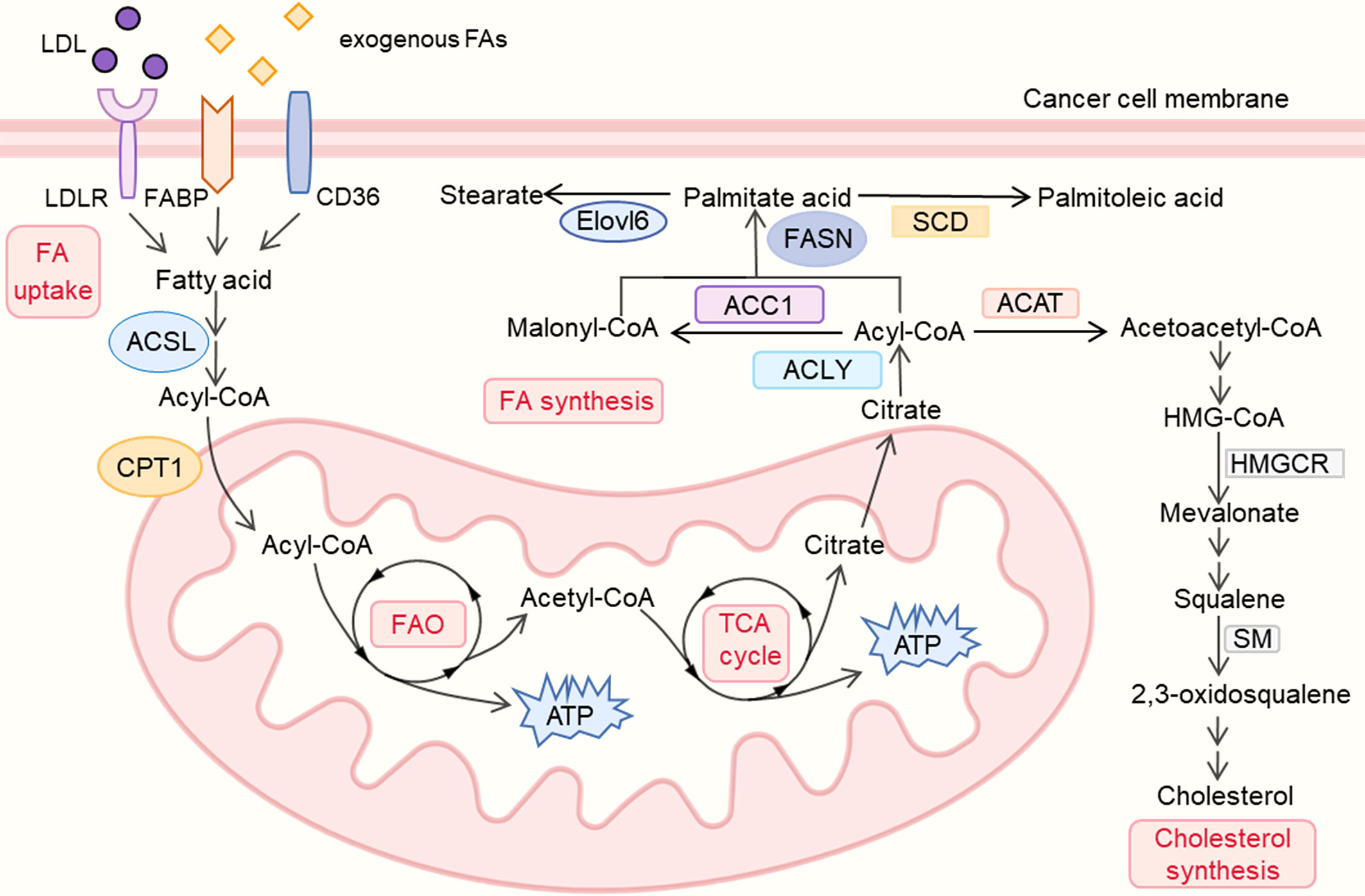
Figure 1. Lipid metabolism reprogramming in cancer cells.
In addition to supporting tumor development, lipid metabolism reprogramming also alters the TME by affecting the recruitment, activation, and function of immune and stromal cells. On the one hand, tumor cells can actively modify the TME by secreting signaling molecules and metabolites, affecting the functions of cancer-associated fibroblasts (CAFs) and immune cells in the TME. On the other hand, lipid metabolic reprogramming, i.e., adaptive changes in cells within the TME manifested by increased lipid uptake and accumulation, or FAO, can drive the TME toward an immunosuppressive phenotype that supports tumor progression. For example, upregulated lipid uptake and FAO increase the lipid metabolism levels of regulatory T cells (Tregs), tumor-associated macrophages (TAMs), and myeloid-derived suppressor cells (MDSCs), promoting their immunosuppressive functions. In addition, the upregulation of CD36 in CD8 + T cells leads to excessive accumulation of lipids, which affects the secretion of anti-tumor factors such as IFN-γ, TNF-α, and ultimately inhibits their anti-tumor effects. Similarly, the upregulation of CD36 in natural killer cells (NK) can also weaken their tumor-killing activity through intracellular lipid accumulation. Studies have shown that blocking lipid uptake by inhibiting CD36 of cytotoxic CD8+ T cells or Tregs enhances anti-tumor immune responses.
Given the critical role of lipids in cancer progression, targeting lipid metabolism-related pathways provides new therapeutic opportunities for cancer. A large amount of evidence shows that inhibitors targeting tumor cell lipid uptake, adipogenesis, and FAO show significant therapeutic effects in various cancers. In addition, regulating lipid metabolism in stromal cells and immune cells also provides new options for anti-tumor treatment. In addition, it can be combined with chemotherapy and immunotherapy, providing a new comprehensive strategy for optimizing cancer treatment.
Current Status and Mechanisms of Lipid Metabolism Reprogramming in Cancer
Lipid Metabolism Reprogramming in Cancer
The majority of lipid molecules in the human diet are triacylglycerols (TAGs) and cholesterol. After TAGs are absorbed, they can be hydrolyzed into glycerol and fatty acids (FAs). Glycerol is then converted into glycerol-3-phosphate (G-3P), which enters the glycolysis process. FAs can either be stored as the main component of membrane synthesis or converted into acyl-CoA to provide energy for β-oxidation. In tumors, several steps of lipid metabolism are generally enhanced to sustain their biological progression. This includes increases in fat absorption, synthesis, storage and FAO.
Carcinogenic Factors Affecting Tumor Lipid Metabolism
Activation of oncogenes and loss of tumor suppressor gene function are the main causes of tumorigenesis. They also play an important role in the reprogramming of tumor metabolism by regulating the expression of lipid metabolism enzymes. Sterol regulatory element binding proteins (SREBPs) are key upstream regulators of lipid metabolism. SREBP is a transcription factor that promotes DNL by upregulating key enzymes such as ACLY, FASN, and SCD. These key enzymes are closely related to tumor proliferation, apoptosis, and invasion. Furthermore, SREBP maintains intracellular cholesterol levels by inducing LDL receptor-mediated cholesterol uptake and inhibiting abca1-mediated cholesterol export in an mTORC1-dependent manner. Mutations in SREBPs and oncogenes (such as PI3K and MYC) and tumor suppressor genes (such as p53 and PTEN) can induce downstream lipid reprogramming events.
Microenvironmental Factors Affecting Tumor Lipid Metabolism
Metabolic reprogramming of cancer cells is the result of a multifactorial process. The TME also plays a crucial role with the activation of oncogenic signals caused by mutations in tumor cells. The TME contains multiple factors, such as hypoxia, acidosis, and malnutrition, which promote tumor occurrence and progression by changing the lipid metabolism of tumor cells.
Current Status and Mechanisms of Lipid Metabolism Reprogramming in TME
As cancer progresses, the TME also undergoes reprogramming of lipid metabolism. It is worth noting that tumor cells play an important role in changing the TME (such as acidosis, lipid accumulation) by producing metabolites and lipid-related signaling molecules. This in turn affects the metabolic pattern and immune phenotype of TME cells, leading to remodeling of the immune microenvironment. For example, CAFs secrete lipids in the TME to directly provide an energy source for tumor cells, thus promoting tumor progression. In addition, the lipid metabolism reprogramming of CAFs also affects their own cytokine secretion function, thereby regulating immune responses and promoting the formation of an immunosuppressive microenvironment. In addition, changes in the lipid metabolism pattern of immune cells are also conducive to the construction of an immunosuppressive microenvironment and support tumor immune escape. Therefore, tumor progression is the result of a co-evolutionary process between the tumor and the TME.