
A study led by stem cell scientists from Harvard University points to a potential new biomarker and drug target which is for amyotrophic lateral sclerosis (ALS) diagnosis. ALS is a neurological disease that is very difficult to diagnose and treat. The study, published in Nature Neuroscience, uses the stem cell model of human motor neurons to reveal the STMN2 gene as a potential therapeutic target, demonstrating the value of this human stem cell model approach in drug discovery.
Diagnosis and treatment of ALS
Motor neuron loss and progressive paralysis will occur in patients with ALS. After a long diagnostic process, they may survive for 5 years. The US Food and Drug Administration (FDA) has approved two ALS drugs, but they only slow down the disease. In addition to effective treatment for more patients with ALS, there is an urgent need for robust testing of ALS. To achieve this, scientists need to find a reliable biomarker for this disease.
TDP-43: biomarker for ALS
About 10 years ago, scientists discovered a protein aggregate called TDP-43 in post-mortem neurons of patients with ALS. This protein should have been found in the nucleus of these neurons, but it was flushed out and accumulated in the cytoplasm (Figure 1). It is obvious that the interaction of some genes in the neuron waste treatment system (called the proteasome) with TDP-43 leads to ALS. However, it is not known which genes are involved and what they are doing. The gene encoding TDP-43 is mutated to trigger ALS. It is passed on to offspring, then developing into amyotrophic lateral sclerosis or, in some cases, developing into frontotemporal dementia (FTD). Since the discovery of TDP-43 aggregates in patients with ALS, TDP-43 has been considered a biomarker of the disease.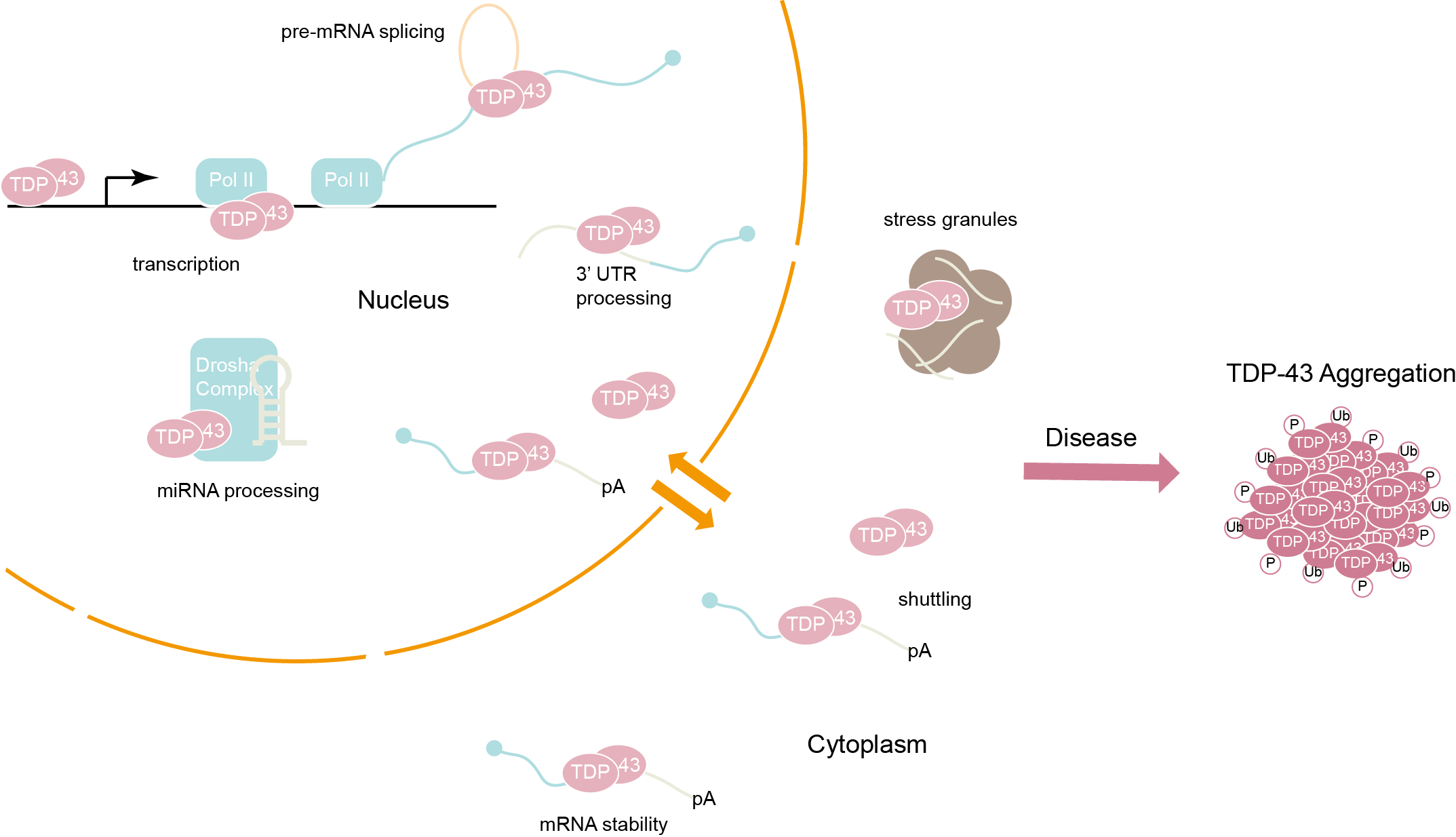
Figure 1. TDP-43 function is presumably compromised upon formation of the inclusion bodies because accumulation of these cytoplasmic aggregates in patients is accompanied by nuclear clearance of the protein.
What are researchers doing and found?
TDP-43 is one of many proteins that bind to RNA, which is responsible for transmitting genetic information and transforming it into specific proteins, such as those required for the growth of neurons. For the first time, the researchers identified all possible RNA types regulated by TDP-43 protein in human neurons. So far, similar studies have only been conducted in mice and cancer cell lines. Then they studied what happened to each gene when they manipulated TDP-43.
The researchers knocked down the levels of TDP-43 protein in motor neurons from human stem cells. Then, using RNA sequencing, they analyzed changes in gene expression in these cells. About 1000 genes were altered when TDP-43 was manipulated, and one of the genes was prominent: Stathmin2 (STMN2), a gene that plays an important role in nerve growth and repair.
They observed that without TDP-43, STMN2’s perfect protein manufacturing instructions would become meaningless. “We found that when the level of TDP-43 in the nucleus was reduced, a mysterious exon was spliced into the STMN2 mRNA, which essentially deleted its instructions for producing functional proteins,” a researcher explained. “This leads to the deletion of the STMN2 protein and thus loses the components required for motor neuron axon repair or growth.
The next step is to see if their findings truly reflect the patient’s biological characteristics. They used data from RNA sequencing studies of autopsy samples from patients with ALS. These rare data sets correspond to the original findings of the research team in the human stem cell model compared to the control group. Data from the spinal cord of patients with ALS reflect the presence of hidden exons, while not found in the control group.
Why is this important?
These experiments make us consider whether ALS can be slowed or stopped if patients repair their STMN2 gene. In other words, these findings suggest a clear way to develop potential treatments for ALS, which is suitable for everyone except very few individuals and is regardless of the genetic cause of their disease.